Физическая химия полимерных гелей Лекция 11 ПОЛИМЕРНЫЙ РАСТВОР, ЭТО ПОВЕРХНОСТНЫЙ СЛОЙ?

Физическая химия полимерных гелей Лекция 10 ПАРАДОКС ШРЕДЕРА. ПРИЧИНЫ РАЗЛИЧНОГО НАБУХАНИЯ ПОЛИМЕРОВ В ПАРЕ И В ЖИДКОСТИ
18.12.2015Физическая химия полимерных гелей Лекция 12 ОСНОВНЫЕ МЕТОДЫ ИЗУЧЕНИЯ СВОЙСТВ И СОСТАВА ПОЛИМЕРНЫХ ГЕЛЕЙ.
18.12.2015Физическая химия полимерных гелей Лекция 11 ПОЛИМЕРНЫЙ РАСТВОР, ЭТО ПОВЕРХНОСТНЫЙ СЛОЙ?

Физическая химия полимерных гелей
Ферапонтов Н. Б., Гагарин А. Н., Струсовская Н. Л., Токмачёв М. Г., Тробов Х.Т., Рубин Ф.Ф.
Москва, МГУ имени М. В. Ломоносова 2015 г.
В работах [1, 2] для описания свойств полимерного геля (ПГ) предложена гетерофазная модель его строения (см. лекцию 7). Если существование фазы внешнего раствора (ВР) в этой модели вполне предсказуемо и экспериментально легко проверяется [3, 4], то причина существования фазы полимерного раствора (ПР) и ее свойства требуют дополнительных обоснований.
С точки зрения коллоидной химии, ПГ следует отнести к связнодисперсным системам, к которым относятся гели и студни [5]. В то же время, ПГ является эмульсией, так как обе его фазы обладают свойствами жидкости. Из сказанного следует, что ПГ являются лиофильными связнодисперсными системами. Такие системы термодинамически устойчивы (DG ? 0) и для них характерно самопроизвольное диспергирование, которое возможно при условии, что возрастание свободной энергии Гиббса DG , связанное с увеличением поверхности при диспергировании, компенсируется уменьшением энтальпии в процессе сольватации и роста энтропии системы за счет поступательного движения образующихся частиц.
Характерная особенность ПР состоит в том, что в отличие даже от мицелл, в которых есть ядро, в ПР часть, называемая объемом, полностью отсутствует. То есть в ПР все полярные группы находятся в поверхностном слое. В связи с этим возникает вопрос: можно ли считать ПР фазой? Напомним, что: фаза, это гомогенная часть системы, однородная по свойствам во всех точках в отсутствие полей. Неоднородность фазы имеет микроскопические масштабы и даже в малом элементе объема ею можно всегда пренебречь. В свое время Гугенгейм назвал поверхностный слой – «поверхностной фазой».
Как уже говорилось, ПР образуется уже при контакте полимера с водяным паром. Известно, что в этом случае полимерная цепь равномерно распределена в объеме набухшего в паре полимера, полярные группы расположены регулярно вдоль полимерной цепи, а количество сорбированной ими воды, иначе говоря, концентрация полярных групп, не зависит от того, в какой части объема полимера или полимерной цепи эти группы находятся [6, 7]. Из сказанного следует, что макроскопические параметры гидратированной полимерной цепи одинаковы по всему объему ПР. К этому следует добавить, что: во-первых, вода объединяет гидратированные полярные группы в один общий объем, а во-вторых при помещении набухшего в паре полимера в раствор, состав ПР изменяется в соответствии с составом внешнего раствора, но сам ПР как фаза, сохраняется, т.е. его состав отличен от состава внешнего раствора. Следовательно, отвечая на вопросы: является ли ПР фазой, и обладает ли ПР свойствами поверхностного слоя, следует ответить положительно.
Таким образом, анализ имеющейся информации о свойствах ПР подтверждает формулировку Гугенгейма о том, что ПР является «поверхностной фазой» т.е. для гидратированных полимерных цепей гидрофильных полимеров характерны свойства как фазы, так и поверхностного слоя.
1. Причины устойчивого существования фазы полимерного раствора.
Свойства всех систем изменяются вблизи поверхности раздела фаз (см.
лекцию 9). Особенно существенны изменения свойств жидкости вблизи твердой поверхности. В результате адсорбции на твердой поверхности происходит изменение концентрации в поверхностном слое относительно концентрации в объеме фазы. Несмотря на малую сжимаемость жидкостей, изменение плотности иногда достигает десятков процентов и распространяется на расстояния нескольких молекулярных диаметров [8]. В граничных слоях структура жидкостей изменяется по сравнению с объемной и тем сильнее, чем полярнее жидкость.
В зависимости от природы действующих сил различают физическую адсорбцию и хемосорбцию. Физическая адсорбция и хемосорбция термодинамически неразличимы, однако практически в большинстве случаев они характеризуются различными значениями дифференциальной молярной теплоты адсорбции:
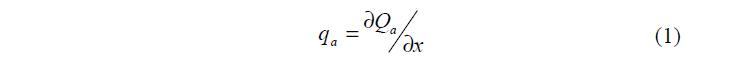
Значения qa для физической адсорбции лежат в пределах 4 – 40 кДж/моль (1 – 10 ккал/моль), а для хемосорбции 40 – 400 кДж/моль (10 – 100 ккал/моль). Зависимость дифференциальной молярной теплоты адсорбции от температуры и давления выражается уравнением:
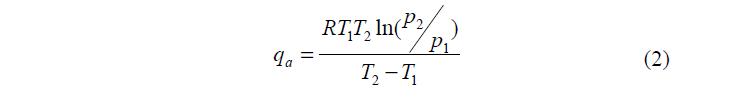
«Установлено, что при адсорбции паров поверхностный слой адсорбата представляет собой жидкость, обладающую особыми свойствами» [5 с. 113].
Cледовательно, процесс адсорбции пара слагается как бы из двух процессов: конденсации пара в обычную жидкость с выделением qL и взаимодействии этой жидкости с адсорбентом, превращением ее в «необычную» жидкость в силовом поле твердой фазы с выделением qx т.е. смачивания. Так как в реальных условиях оба процесса протекают одновременно, то:
![]()
При контакте полярного адсорбента с полярной жидкостью, теплоты смачивания, отражающие энергию взаимодействия твердого тела с жидкостью, обычно велики.
Ионы в растворе являются носителями электрических зарядов, поэтому переход ионов из объема фазы в поверхностный слой сопровождается перераспределением зарядов и возникновением электрического поля в области поверхностного слоя. При этом в поверхностном слое образуется двойной электрический слой (ДЭС).
Полиэлектролиты от обычных НМС отличаются наличием пространственного электрического заряда, характеризующегося высокой плотностью вдоль молекулы и в ее ближайшей окрестности. Это связано с тем, что расстояния между фиксированными группами полиэлектролита не изменяются при растворении. По этой причине пространственный заряд не зависит от концентрации полиэлектролита так как для соблюдения условий электронейтральности необходимо, чтобы концентрация противоионов, образующих пространственный заряд, равнялась концентрации фиксированных ионов. В окрестности фиксированных ионов полимерной молекулы противоионы образуют ионную атмосферу [9].
Для термодинамического рассмотрения процессов переноса заряженных частиц, необходимо ввести в фундаментальные уравнения еще один член, выражающий электрическую компоненту энергии. Этот член равен произведению заряда q на потенциал j , имеющий размерность энергии. Для молярной энергии Гиббса он равен z Fj i . Условия равновесия выражаются в этом случае (при постоянных Р и Т) равенством не химических, а электрохимических потенциалов:
![]()
В соприкасающихся фазах 1 и 2
![]()
При контакте фаз происходит переход ионов одного вида из фазы с большим значением i m в фазу с меньшим значением. Этот процесс приводит к возникновению межфазного скачка потенциала 2 1 Dj =j -j , препятствующему дальнейшему переходу ионов. В результате устанавливается равновесие, которому отвечает равновесный скачок потенциала, равный:
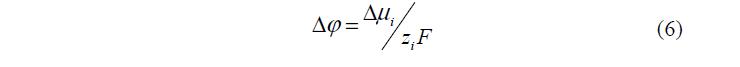
где: 2 1 i i i Dm = m — m Таким образом, в состоянии равновесия фазы заряжаются разноименно и возникает ДЭС [10]. В области ДЭС равновесные концентрации ионов отличаются от таковых в объеме раствора и возникают поверхностные избытки заряженных компонентов.
2. Влияние диффузного электрического слоя на свойства полимерного раствора. Связь между электрохимическим потенциалом и константой распределения.
Существование ДЭС вдоль полимерной молекулы должно приводить к тому, что активности компонентов в нем должны отличаться от активностей в остальном объеме. Этот факт был экспериментально установлен и описан в работе [11]. Там же было показано, что отношение активностей компонентов в ДЭС и равновесном растворе есть величина постоянная, не зависящая от концентрации ВР. На этом основании в гетерофазной модели были введены константы распределения компонентов между фазами ПР и ВР.
Как известно из электрохимии, равновесие в системе, в которой контактирующие фазы имеют разные электрические потенциалы j , определяется равенством не химических (7),
![]()
а электрохимических потенциалов компонентов i m~ , связанных с их химическими потенциалами i m соотношением:
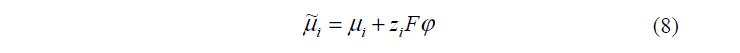
где: zi – зарядовое число иона; j – потенциал в ДЭС; A F = eN – число Фарадея.
Для каждого вещества условию равновесия в этом случае отвечает постоянное значение электрохимического потенциала по всей системе, т.е.
![]()
Такая запись учитывает три основных фактора, определяющих поведение ионов в системе: их молекулярное взаимодействие с окружающей средой io m , участие в тепловом движении RT ln ni и взаимодействие с электрическим полем z Fj i . Соотношение (9) должно выполняться для всех ионов, присутствующих в системе. Очевидно также, что в соответствии с уравнением Гиббса-Дюгема, в этом случае и активность воды в фазе ПР также будет отличаться от активности воды во ВР.
При заданной температуре и заданном давлении, величина электрического потенциала, однозначно определяется природой полимера и составом раствора, т.е. соответствующими активностями. Как следует из уравнений (7-9) связь между константой распределения и электрическим потенциалом полимерной цепи можно записать:
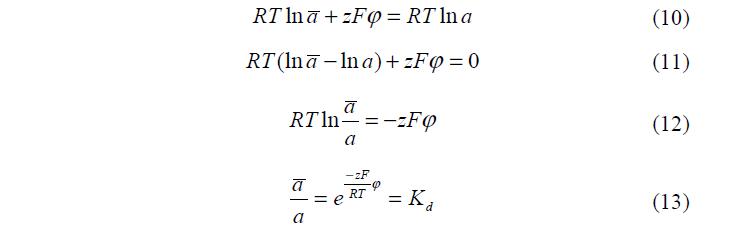
Уравнение (13) показывает связь между электрическим потенциалом полимера и константой распределения вещества в фазах ПГ.
ДОПОЛНИТЕЛЬНЫЙ МАТЕРИАЛ.
3. Теория Гуи – Чепмена.
Основана на идее подвижности ионов внешней обкладки (противоионов) [5]. Электростатическое (кулоновское) притяжение их к поверхности (к внутренней обкладке) и отталкивание коионов – ионов, заряженных одноименно с поверхностью, уравновешивается тепловым движением ионов (диффузией) размывающим поверхностные избытки. Слой раствора с измененными концентрациями ионов вблизи поверхности называется диффузным. В зависимости от условий его толщина изменяется от нм до сотен мкм.
Теоретическая обработка этой модели позволила установить связь между зарядом r , потенциалом j и равновесной концентрацией электролита в растворе с. Для потенциала в диффузном слое можно записать следующие граничные условия: при х = ?, j = 0 и = 0 dx dj при 0 = х , 0 j j = Условие электронейтральности ДЭС записывается в виде:
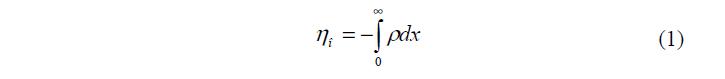
где: 0 h – плотность поверхностного заряда; r – объемная плотность заряда в растворе (Кл/м3); х – расстояние.
Заряд r складывается из избытка противоионов (сi >coi) и недостатка коионов (c-<coi) в растворе:

В каждой точке он фактически равен разности (сi+ – ci) (поскольку zi имеют разные знаки), умноженной на zF (коэффициент перехода от химических единиц к электрическим). Концентрации сi противоионов и коионов в растворе вблизи поверхности определяются законом Больцмана:
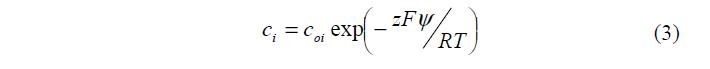
Потенциал j связан с зарядом r уравнением Пуассона
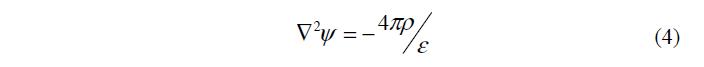
где p – поверхностное давление; e – диэлектрическая проницаемость.
Для плоского ДЭС уравнение (4) принимает вид:
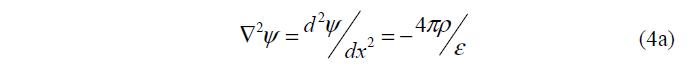
Подстановка (2) и (3) в (4) дает уравнение Пуассона – Больцмана:
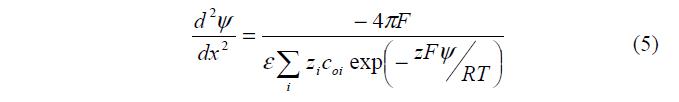
4. Некоторые следствия теории диффузного электрического слоя.
Представления о ДЭС, как о плоском конденсаторе, позволяет по величине емкости оценить его толщину (протяженность в жидкой фазе).
В плотном слое dx const dy = , следовательно:
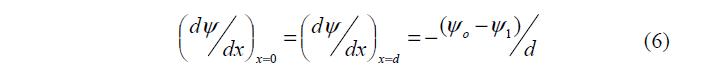
где: 1 y – потенциал плоскости наибольшего приближения.
Подстановка (4а) в (1) с последующим интегрированием и учетом (6) дает:
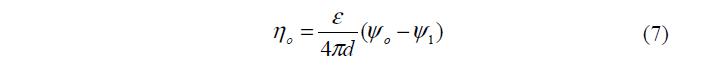
Сопоставляя с известной формулой для плоского конденсатора h Сj о = , находим, что 1 j =y -y о , а емкость плотного слоя:
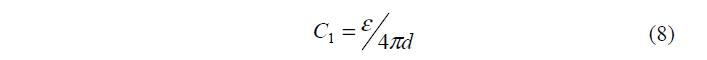
Кроме того, нетрудно установить, что при с ®0 о y ®y 1 .
Для нахождения емкости слоя в случае малых потенциалов можно получить:
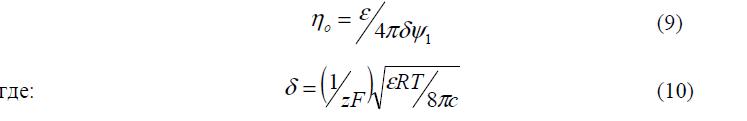
Поскольку (9) идентично формуле для плоского конденсатора, параметр d получил название приведенной толщины диффузного слоя.
Таким образом, при малых 1 y заряд о h и потенциал 1 y оказываются прямо пропорциональными. При этом двойной слой ведет себя как плоский конденсатор с расстоянием между пластинами, равным d , емкость которого, равна емкости реального диффузного слоя. Это означает, что реальный диффузный слой эквивалентен такой модели, в которой все противоионы отстоят на одинаковом расстоянии d от плоскости х=d.
Из уравнения (10) следует, что d не зависит ни от плотности поверхностного заряда о h , ни от потенциала ( о j или 1 j ), а является функцией лишь заряда ионов и концентрации электролита (при Т=const).
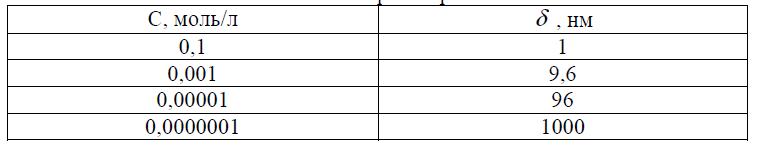
Величинаd уменьшается линейно с ростом с . Расчет по (10) при z=1 и T=300K дает конкретные значения приведенной толщины диффузного слоя d в водных растворах. Как следует из таблицы, диффузный слой в разбавленных растворах может простираться на расстояния порядка сотен ионных радиусов.
Таблица 1. Зависимость толщины диффузного слоя (ДС) от концентрации внешнего раствора.
ЛИТЕРАТУРА.
1. Ферапонтов Н.Б. Горшков В.И., Тробов Х.Т., Парбузина Л.Р. Изучение равновесия ионит раствор на примере сульфокатионита КУ-2. // Журн. физ.химии. 1994. Т. 68, 6. C. 1109-1113.
2. N.B. Ferapontov, V.I. Gorshkov, L.R. Parbuzina, H.T. Trobov, N.L. Strusovskaya. React. & Funct. Polym. 41 (1999) 213.
3. Х.Т. Тробов Равновесие между моноионными формами ионитов и растворами простых электролитов // Дисс… канд. хим. наук.. М. МГУ, 1994, 141с.
4. Архангельский Л.К. Матерова Е.А. О некоторых закономерностях поглощения паров воды смешанными формами сульфокатионитов с различным числом поперечных связей // Вестник ЛГУ.. 1968. Т. 10, 2. C. 146-148.
5. Фридрихсберг Д.А. «Курс коллоидной химии» Ленинград. Химия. 1984. 368с.
6. Полянский Н.Г., Горбунов Г.В., Полянская Н.Л. «Методы исследования ионитов». // М.: Химия., 1976, 208с.
7. Даванков В.А., Цюрупа М.П. Сверхсшитый полистирол – новый тип сорбентов //Итоги науки и техники. 1984. Т.5. С. 32.
8. Дерягин Б.В., Чураев Н.В. «Surface and Membrane Scince», 1981 v.14, p. 69-130.
9. Корыта И. «Ионы, электроды, мембраны».(Мир. Москва, 1983), 264с.
10.Дамаскин Б.Б., Петрий О. А. «Введение в электрохимическую кинетику». М.: Высшая школа, 1976. 416с.
11.Ferapontov N.B., Parbuzina L.R., Gorshkov V.I., Strusovskaya N.L., Gagarin A.N. Interaction of cross-linked polyelectrolytes with solutions of low-molecular-weight electrolytes.// Reactive & Functional Polymers. 2000. V.45. P. 145-153.