Физическая химия полимерных гелей Лекция 20 ГЕТЕРОФАЗНАЯ ФИЗИКО-МАТЕМАТИЧЕСКАЯ КИНЕТИЧЕСКАЯ МОДЕЛЬ НАБУХАНИЯ ПОЛИМЕРНЫХ ГЕЛЕЙ
Физическая химия полимерных гелей Лекция 23 РАЗВИТИЕ ПРЕДСТАВЛЕНИЙ О КИНЕТИКЕ НАБУХАНИЯ ПГ
19.12.2015
Физическая химия полимерных гелей Лекция 21 ВЛИЯНИЕ РАЗМЕРА ГРАНУЛ И КОНЦЕНТРАЦИИ РАСТВОРА KCL НА КИНЕТИКУ НАБУХАНИЯ ИОНИТОВ: КУ-2Х4, КУ-2Х8, АРА4П И АВ- 17Х8
19.12.2015Физическая химия полимерных гелей Лекция 20 ГЕТЕРОФАЗНАЯ ФИЗИКО-МАТЕМАТИЧЕСКАЯ КИНЕТИЧЕСКАЯ МОДЕЛЬ НАБУХАНИЯ ПОЛИМЕРНЫХ ГЕЛЕЙ

Физическая химия полимерных гелей
Ферапонтов Н. Б., Гагарин А. Н., Струсовская Н. Л., Токмачёв М. Г.
Москва, МГУ имени М. В. Ломоносова 2015 г.
В основу физико-математической кинетической модели набухания ПГ [1] заложена гетерофазная модель, выделяющая в набухшем полимере две фазы: фазу полимерного раствора (ПР) и фазу внешнего раствора (ВР) [2–4].
Гетерофазная физико-математическая, кинетическая модель набухания полимеров представлена в форме системы уравнений, решение которой позволяет получить изменение степени набухания полимера во времени, определить время релаксации системы и равновесные значения степени набухания. Система уравнений апробирована на гранулах сферической формы. Влияние других форм образцов полимеров на кинетику набухания пока не исследовано. Кроме того, в модели не рассматриваются случаи, сопровождающиеся ионным обменом и случаи, когда растворенное вещество проникает в фазу ПР. Схема кинетической гетерофазной модели приведена на рис.1.
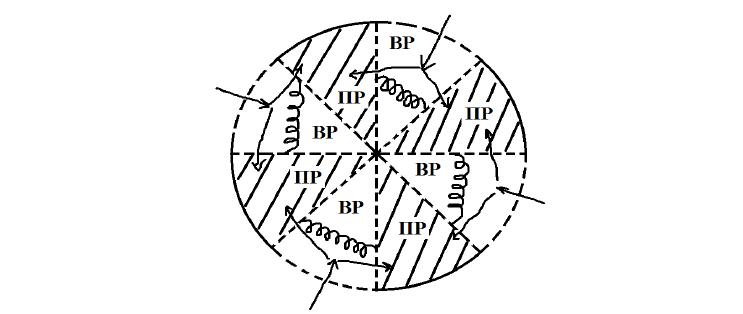
Рис.1. Кинетическая схема двухфазного строения набухшего полимера – полимерного геля (ПГ). ПР – фаза полимерного раствора, состоящая из полимера и «связанной» воды.
Объем этой фазы изменяется в зависимости от активности воды в растворе снаружи; ВР – фаза внешнего раствора или «свободной» воды. При равновесии состав этой фазы не отличается от состава раствора снаружи.
Удобство применения гетерофазной модели для описания кинетики набухания ПГ заключается в возможности использования при проведении расчетов справочных данных о свойствах компонентов системы, к которым относятся основные свойства полимеров и растворов. Кроме таких данных в систему уравнений входят три коэффициента: k1 [м/с] – характеризует скорость потока из гранулы (или в гранулу) под воздействием градиента химического потенциала воды (разность активностей воды); k2 [с-1] – отражает характерное время релаксации полимерного каркаса к положению равновесия; k3 @D/R0 [м/с] – показывает скорость проникновения растворенного вещества внутрь гранулы под воздействием градиента химического потенциала воды (разность концентраций).
Если значения указанных коэффициентов известны, то для любой концентрации раствора в данной системе время релаксации и изменение степени набухания ПГ могут быть рассчитаны без проведения эксперимента.
1. Описание кинетики набухания полимерного геля.
В качестве примера рассмотрим кинетику изменения степени набухания гранулы ПГ сферической формы при переносе ее из воды в раствор. В находящемся в жидкости полимере, часть объема ПГ и, соответственно, его поверхности занимает ПР (рис. 1), в котором, в соответствии с константой распределения воды w dist K , активность «связанной» воды меньше, чем активность «свободной» воды, находящейся в ПГ в фазе ВР (уравнение I ) [3].
При этом количество «связанной» воды в фазе ПР максимальное, т.е.
соответствует максимальному значению на изотерме сорбции воды данным полимером (рис.2).
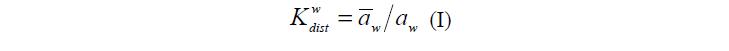
где w a и w a – активности воды в фазах ПР и ВР соответственно.
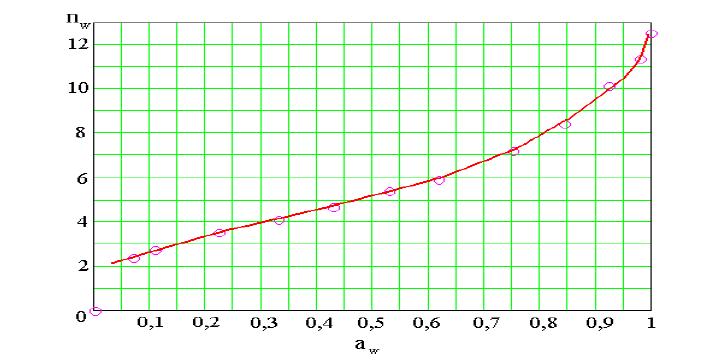
Рис. 2. Пример изотермы сорбции воды ионитом.
Для проведения вычислений вначале определяют объём занимаемый фазой ВР в ПГ. Его нетрудно найти, если известны объемы ПГ и фазы ПР (уравнение II).
![]()
где eq s V – максимальное количество «связанной» воды в фазе ПР определяемое по изотерме сорбции воды данным полимером (при w a =1).
В растворе, в который помещают полимер, активность воды всегда меньше, чем в чистой воде, поэтому переход ПГ от равновесия вода – ПГ к равновесию раствор–ПГ, в соответствии с изотермой сорбции воды, сопровождается уменьшением количества «связанной» воды в фазе ПР (рис.2).
Уменьшение количества «связанной» воды в фазе ПР приводит к уменьшению диаметра гидратной оболочки вокруг полимерной цепи, в результате чего объем, занимаемый полимерной сеткой, уменьшается. Это и есть причина уменьшения объема всего ПГ. Перешедшая из фазы ПР вода, оказывается за пределами ПГ и образует вокруг него приповерхностный слой (рис.3).
Отметим, что в некоторых случаях вместе с уменьшением объема ПР уменьшается объем и ВР. Вся вода, оказавшаяся за пределами ПГ, перемешивается с внешним раствором и тем самым снижает его концентрацию вблизи поверхности гранулы (рис. 4), что отражается на кинетике набухания.
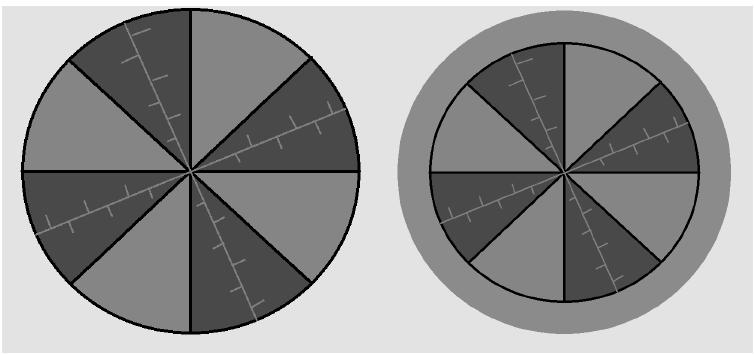
Рис. 3. Образование приповерхностного слоя в результате уменьшения размера гранулы.
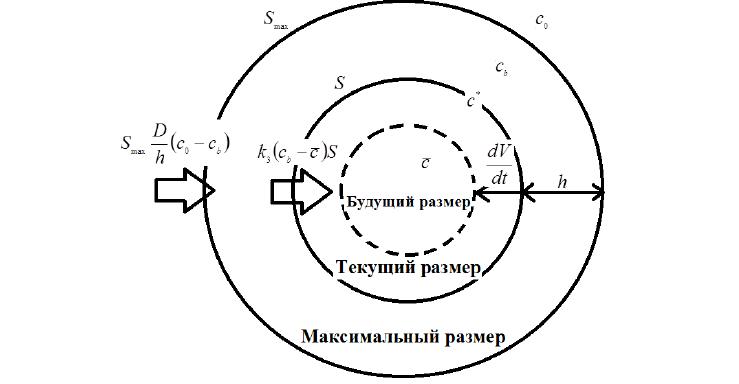
Рис. 4. Схема распределения концентраций раствора внутри и в приповерхностном слое гранулы в некоторый момент времени ti . Smax – максимальное значение площади видимой поверхности гранулы в эксперименте (чаще всего это размер гранулы в воде), S – текущая площадь поверхности гранулы; h – текущая толщина приповерхностного слоя.
Другую часть, как объема, так и поверхности ПГ занимает фаза ВР (см.
рис.1). Для полимеров, на основе полистирола, сшитого дивинилбензолом объём этой фазы не меняется при изменении концентрации внешнего раствора [5]. Из фазы ВР «свободная» вода вытесняется за пределы ПГ раствором, находящимся снаружи ПГ. Скорость вытеснения зависит от разности химических потенциалов (активностей) воды в вытесняемой и вытесняющей фазах, а также от плотности и вязкости внешнего раствора. Ниже приведено математическое описание процесса изменения степени набухания ПГ, происходящее в результате изменения состава внешнего раствора.
Пусть ПГ изначально находится в воде. В этом случае фазу ВР в ПГ заполняет «свободная» вода. При помещении гранулы в раствор, разность химических потенциалов «свободной» воды в ПГ и воды в растворе снаружи создает поток воды из ПГ, точнее из ПР, во внешний раствор (первое слагаемое правой части уравнения (1)). На скорость потока воды кроме разности электрохимических потенциалов, а проще активностей воды, оказывает влияние упругость полимерной матрицы (второе слагаемое правой части уравнения (1)).
Как уже говорилось выше, в результате сжатия полимерного каркаса вокруг него снаружи образуется приповерхностный слой, изменение концентрации в котором описывает уравнение (2).
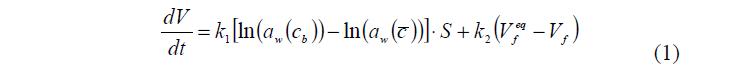
где bound b c = c – концентрация растворенного вещества в приповерхностном слое. Ее можно определить из условия равенства потоков:
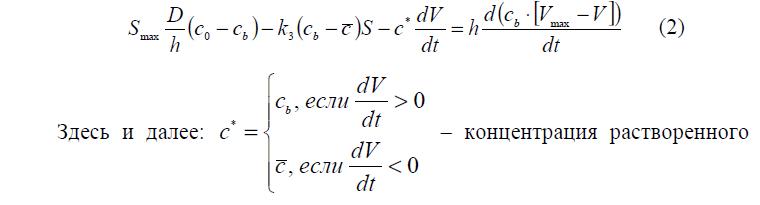
Здесь и далее: концентрация растворенного вещества непосредственно на видимой поверхности гранулы.
В отличие от традиционного осмотического процесса, поток воды из ПГ сопровождается образованием альтернативного потока внешнего раствора внутрь ПГ. Это приводит к появлению и росту количества и концентрации растворенного вещества в фазе ВР. Скорость увеличения количества растворенного вещества в ПГ зависит от концентрации раствора вблизи поверхности гранулы, которая, в свою очередь, постоянно изменяется из-за вытесняемой из ПГ воды, (см. уравнение (3)).
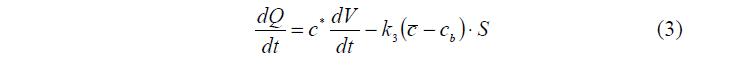
Увеличение концентрации растворенного вещества в фазе ВР приводит к уменьшению количества воды в фазе ПР и, соответственно, объема всего ПГ.
Этот процесс описывается уравнением (4).
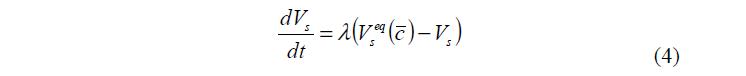
Скорость установления равновесия между фазами внутри гранулы принимали равной ?=104 с–1, что значительно больше скоростей других процессов, поэтому изменение объема фазы ПР происходило без запаздывания. Таким образом, можно считать, что
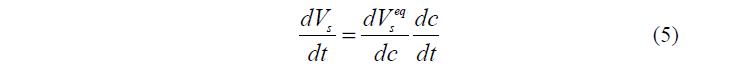
где первый множитель вычисляется на основании данных об изотерме сорбции воды, а второй – согласно уравнению (3).
Для полимеров, типа ПВС, ПААм и некоторых других, замена раствора снаружи сопровождается изменением объема обеих фаз, т.е. не только фазы ПР, но и фазы ВР. В этом случае, для учета изменения объема фазы ВР, в систему уравнений, описывающих кинетику набухания, добавляется уравнение (6)
![]()
где: р [л/моль] – параметр, описывающий равновесную степень набухания данного полимера в растворе данной концентрации.
Использование приведенных выше уравнений с учетом соответствующих начальных условий образуют полную модель процесса.
Таким образом, задав значения коэффициентов k1, k2 и k3 можно построить расчетную кинетическую кривую.
Найти значения коэффициентов k1, k2, k3 на основании данных эксперимента можно, решая обратную задачу. Путем анализа функции ( ) 0 V f t Vi = подбирают такие значения коэффициентов, чтобы расчетная кривая описывала экспериментальную, в пределах ошибки эксперимента (рис.
5). В этом случае, определив значения коэффициентов модели, можно априорно определять конечное изменение степени набухания ПГ и время, необходимое для релаксации ПГ при любой концентрации раствора в изучаемой системе.
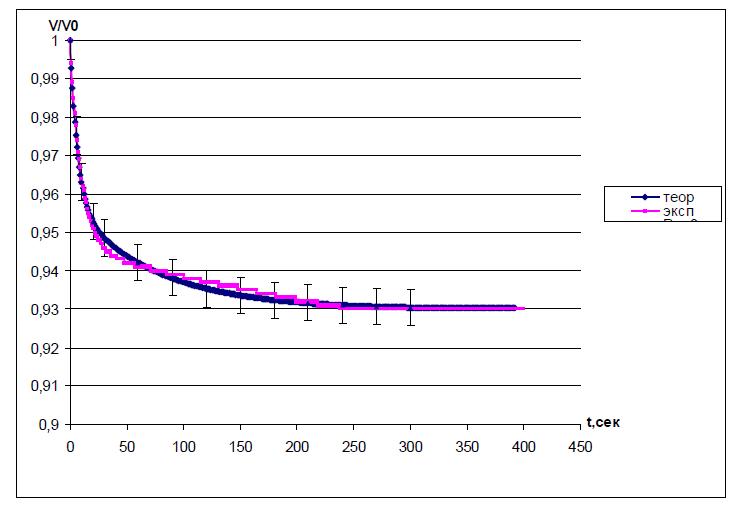
Рис. 5. Аппроксимация экспериментальной кинетической кривой.
ЛИТЕРАТУРА
1. Ферапонтов Н.Б., Токмачев М.Г., Гагарин А.Н., Герасимчук В.В., Пушкарева И.В. Влияние свойств полимеров на условия их набухания в воде и в водных растворах // Сорбционные и хроматографические процессы, 2014, Т. 14, Вып. 5, стр. 703-720.
2. Ферапонтов Н.Б., Горшков В.И., Тробов Х.Т., Парбузина Л.Р. Изучение равновесия ионит-раствор на примере сульфокатионита КУ-2. // Ж. физ.Химии, 1994, Т. 68, № 6, с.1109-1113 .
3. Ferapontov N.B., Gorshkov V.I., Parbuzina L.R., Trobov H.T. et al.Heterophase model of swollen cross-linked polyelectrolyte.// Reactive and functional polymers, 1991, V. 41, p. 213-225.
4. Ferapontov N.B., Parbuzina L.R., Gorshkov V.I., Strusovskaya N.L., Gagarin A.N. Interaction of cross-linked polyelectrolytes with solutions of lowmolecular- weight electrolytes.// Reactive & Functional Polymers, 2000, V. 45, P. 145-153.
5. Ferapontov N.B., Tokmachev M.G., Gagarin A.N., Strusovskaya N.L., Khudyakova S.N. Influence on the environment on swelling of hydrophilic polymers // Reactive and functional polymers, 2013, V. 73, p. 1137-1143.
Список обозначений